(modified last September 18, 2004)
A paper describing this work has been published in the American Journal of Human Genetics (S. White et al. (2002) 71: 365-374).
Although ~95% of deletions can be detected in males using multiplex PCR, other methods must be used to determine duplications, as well as the carrier status of females. The most commonly applied methods are quantitative multiplex PCR and quantitative Southern blotting. The drawback of quantitative multiplex PCR is that often not all exons are examined, meaning that small and rare mutations are missed. Southern blotting is usually applied to determine the boundaries of the mutation, which is essential for distinguishing DMD from BMD, i.e. frame disrupting from open reading frame changes. Using high-quality Southern blots it is possible to perform a quantitative analysis and detect duplications. However, this technique is time consuming, it is difficult to exactly determine the duplication boundaries, it can be difficult to detect duplications in females and triplications will be missed.
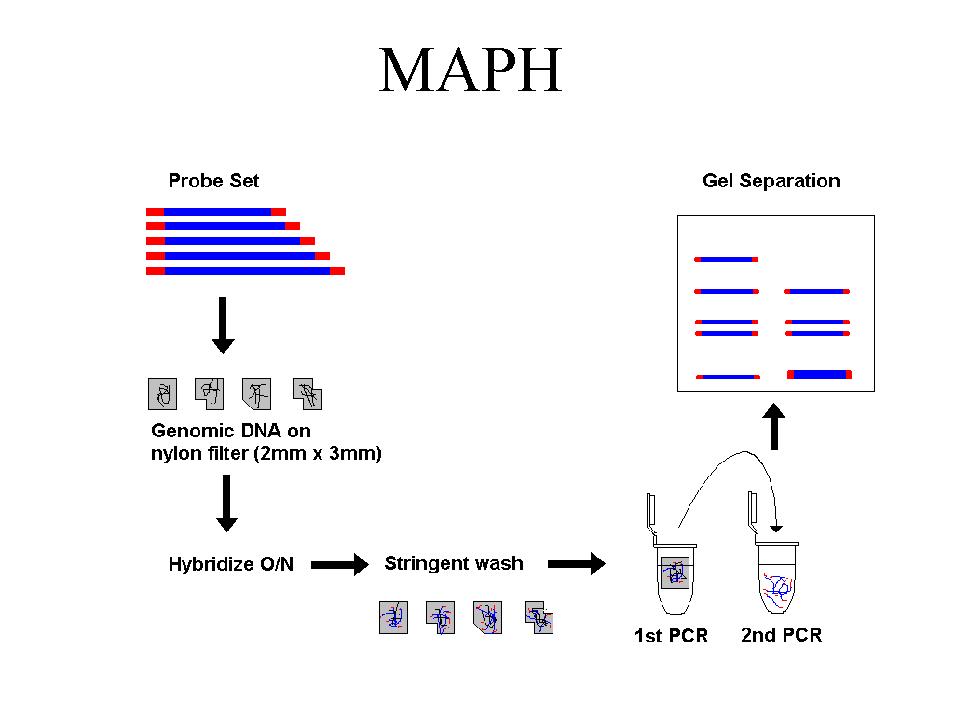
We have developed a system for analysing all 79 exons of the DMD gene for deletions and duplications. It is based on a technique published by Armour et al (Nucl.Acids Res. 2000, 28: 605-609), called Multiplex Amplifiable Probe Hybridisation (MAPH, Fig.1). MAPH is based on a quantitative PCR of short DNA probes recovered after hybridisation to immobilized genomic DNA. Each probe represents a single exon, which is amplified and cloned into a specific vector. By reamplification using primers from the multiple cloning site it possible to co-amplify all probes in one reaction using a single pair of primers.
We divided the exons of the human DMD gene into 2 pools, which are used in separate reactions. 1 ug of denatured genomic DNA is spotted on a small nylon filter, and hybridized overnight in a solution containing one of the probe mixes. Following stringent washing the next day the filter is placed in a PCR tube and a short PCR reaction is performed. This releases the specifically-bound probes into the solution. An aliquot of this is transferred to a second, quantitative PCR reaction.
We choose to analyse the samples by capillary electrophoresis, so one of the primers used in the second PCR must be fluorescently labeled. After PCR the samples are loaded on the ABI3700 (Applied Biosystems), a 96 capillary sequencer. For each sample a series of peaks is generated, with the height of each peak being directly proportional to the copy number of the appropriate exon (Fig.2). By comparing peaks between controls and test samples we have been able to detect single exon deletions and duplications missed using other techniques. We are currently working on further automation of the technique, both in hybridisation and data analysis. Amongst others, analysis of the products on micro-arrays of spotted exons is being tested (Fig.3).
Details regarding availability of the probe set. For those interested we offer deletion / duplication screening of the DMD-gene using both MLPA and MAPH (for information please contact us; Johan den Dunnen - E-mail: ddunnen @ LUMC.nl).
Legend:
Outline of the MAPH technique. The products can be analysed either by separation on a
polyacrylamide gel (as in Armour
et al. [2000]), or by capillary electrophoresis (see Figure 2).
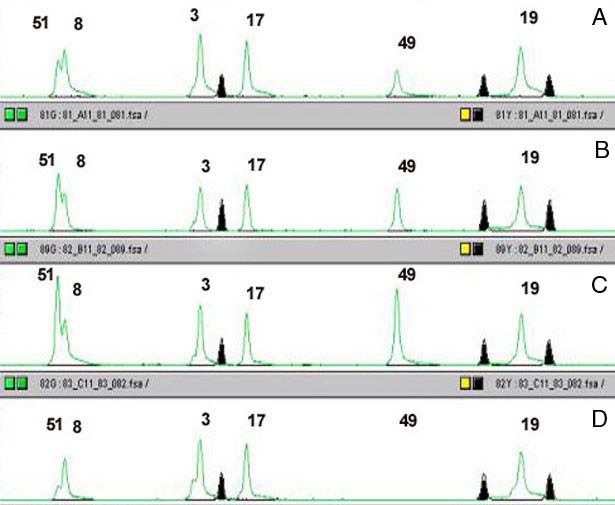
Legend:
Example of the results obtained on a capillary sequencer (ABI 3700).
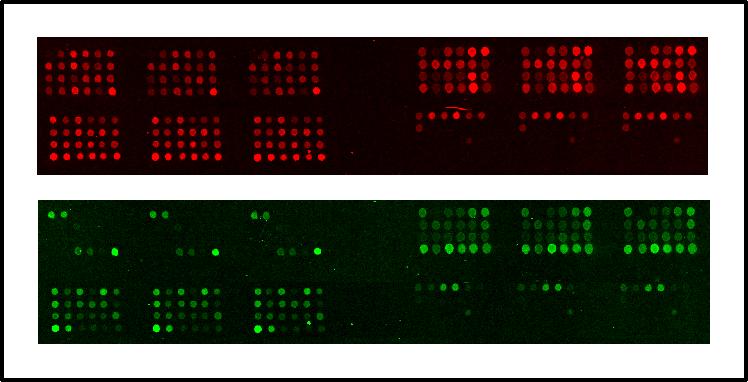
Legend:
Example of the results obtained on a micro-array. PCR-fragments containing DMD exons are
spotted in triplicate on each array (top left exons 1-24, top right exons 25-48,
bottom left exons 49-72, bottom right exons 73-79). Top: signal
obtained with control DNA (Cy-5 labeled). Bottom: signal obtained with
DNA from DMD-patient (Cy-3 labeled), deleted for exons 3-20. The arrays shows that exons
3-20 for the patient DNA do not give a signal, so are deleted.
| Top of page | LMDp homepage | Diagnostic techniques |
| obtaining
the DMD MAPH probe set |
| Remarks / information | Disclaimer
|