(last modified on October 6, 2007)
Dystrophin is a large, rod-like cytoskeletal protein which is found at the inner surface of muscle fibers. Dystrophin is missing in Duchenne Muscular Dystrophy (DMD) patients or reduced in amount in Becker Muscular Dystrophy (BMD) patients. When dystrophin is solubilized from the sarcolemmal fraction it is associated with a large oligomeric complex of sarcolemmal proteins and glycoproteins, the dystrophin-glycoprotein complex (DGC). The proteins in this complex were first described by Campbell and coworkers, designating the components as dystrophin-associated proteins (DAPs) and dystrophin- associated glycoproteins (DAGs). The original names given reflect the approximate molecular weigths: 156DAG, 59DAP, 50DAG, 43DAG, 35DAG and 25DAP. Ozawa and co-workers identified the same complex, referring to the proteins as A0, A1, A2, A3a/A3b, A4 and A5. Later, the proteins were divided into two sub-complexes: the dystroglycan and the sarcoglycan complex (Yoshida).
The DGC forms a critical link between the cytoskeleton and the extra-cellular matrix. The constituents of the DGC differ between different tissues. Little is known regarding the DGC in non-muscular tissues. alpha-Dystroglycan is an extra-cellular protein which binds to laminin-alpha2, probably through its sugar chains, and to beta-dystroglycan. Laminin is a component of the basal lamina (extracellular matrix), comprised of three subunits: alpha, beta and gamma. The laminin-alpha2 (merosin) subunit binds to alpha-dystroglycan. beta-Dystroglycan is a transmembrane protein which, outside the cell, binds to alpha-dystroglycan and, inside the cell, to the cysteine-rich domain and the first half of the carboxyl-terminal domain of dystrophin (together designated the dystroglycan binding [D-]domain). In its N-terminus, dystrophin binds cytoskeletal F-actin. Since alpha- and gamma-sarcoglycan are not expressed in smooth muscle cells and epsilon-sarcoglycan is not an integral component of the skeletal muscle DGC, the association between the SGC-complex and alhpa-dystroglycan is either mediated by beta- and delta-sarcoglycan or by sarcospan (Straub). Yoshida et al. found that the N-terminal half of dystrobrevin participates in an association with the sarcoglycan-sarcospan complex. The authors hypothesized that the sarcoglycan-sarcospan complex is linked to the signaling protein neuronal nitric oxide synthase via alpha-syntrophin associated with dystrobrevin.
The Drosophila DGC has been recently described by Greener and Roberts (for details see The DGC in Invertebrates); it consists of 8 proteins, one dystrophin (DmDys), one dystrobrevin (DmDYB), two syntrophins (DmSYN-1 and DmSYN-2), three sarcoglycans (DmSCG-ae, DmSCG-b and DmSCG-yd) and a dystroglycan (DmDG).
Legend:
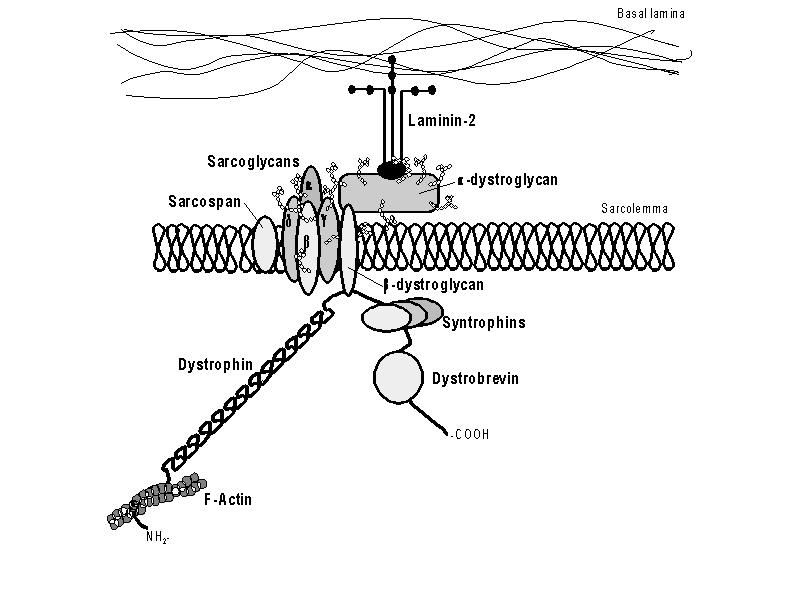
Schematic drawing of the dystrophin complex. NH2- and -COOH indicate the
dystrophin amino- and carboxy-terminus resp. In some tissues, the constitution
of the sarcoglycan complex differs; alpha-sarcoglycan is replaced by
epsilon-sarcoglycan (and gamma-sracoglycan is absent). Other (color) schematic examples
are available from e.g. Louise Anderson (protein
directed, disease
directed).
Integral components of the dystrophin-glycoprotein complex (DGC) are defined by four biochemical and cellular criteria (Crosbie).
Using these criteria, proteins like e.g. caveolin (Crosbie) turn out not to be integral members of the DGC, but proteins whose association is less tight.
The components of the DGC include several proteins on the cytoplasmic side (e.g. dystrophin, the syntrophins and dystrobrevin) and two transmembrane glyco-protein sub-complexes (the dystroglycans and sarcoglycans). The DGC is comprised of:
Other proteins are associated to this complex, but less tightly:
Details on the constituents of the DGC in evolutionary more distant organisms have been described for the fruit fly (Drosophila melanogaster, see The DGC in Invertebrates) and wurm (Caenorhabditis elegans).
Straub et al report an extensive analysis of the DGC of striated (cardiac and skeletal) versus smooth muscle in mouse (embryos, adults, sgca-/-, mdx and mdx/utrn-/-) and the BIO14.6 hamster. The smooth muscle DGC contained dystrophin, alpha- and beta-dystroglycan, beta-, delta- and epsilon-sarcoglycan and sarcospan. In smooth muscle, the molecular weight of delta-sarcoglycan and beta-dystroglycan appeared to be slightly lower. Furthermore, in smooth muscle, two alpha-dystroglycan bands,156 and 100 kDa, were observed (Straub).
Blake found that the composition of the dystrophin-associated protein complex in the brain differs from that in muscle. Because beta-dystrobrevin and dystrophin are expressed in similar populations of neurons in the hippocampus and cortex, it is possible that beta-dystrobrevin interacts directly with dystrophin. If this is the case, then beta-dystrobrevin levels may be reduced in DMD patients similar to the reduction in sarcolemmal staining seen with other components of the DGC in dystrophic muscle. The findings may be relevant to the cognitive dysfunction affecting DMD/BMD patients.
Originally, dystroglycan was described by Smalheiser & Kim as "cranin". Later, Ervasti et al. described the same protein as 156 (alpha-) and 43 kD (Ô-) proteins associated with dystrophin. Ibraghimov-Beskrovnaya et al., who suggested the name "dystroglycan" (dystrophin-associted glycoprotein), showed that both proteins are ubiquitously expressed and encoded by a single chromosome 3p21-derived 5.8 kb transcript. Mature alpha- and Ô-dystroglycan are derived from this transcript by post-translational processing of the encoded 97 kD precursor protein. alpha-dystroglycan contains a single transmembrane domain, three potential N-linked glycosylation sites and a cytoplasmic tail. Ô-dystroglycan contains no transmembrane domain, one potential N-linked and numerous O-linked glycosylation sites. Dystroglycan is heavily glycosylated. The muscle and nonmuscle isoforms of dystroglycan differ by carbohydrate moieties but not protein sequence.
Dystroglycan is the laminin binding component of the dystrophin-glycoprotein complex which provides a link between the subsarcolemmal cytoskeleton and the extracellular matrix. Dystroglycan also functions as dual receptor for agrin and laminin-2 in the Schwann cell membrane. alpha-Dystroglycan is an extra-cellular protein which binds to alpha-laminin (merosin), probably through its sugar chains, and to Ô-dystroglycan. Laminin is a component of the basal lamina (extracellular matrix), comprised of three subunits: alpha, Ô and gamma. Ô-dystroglycan is a transmembrane protein which, outside the cell, binds to alpha-dystroglycan and, inside the cell, to the cysteine-rich domain and the first half of the carboxyl-terminal domain of dystrophin (together designated the dystroglycan binding (D-) domain). No human disease has yet been linked to dystroglycan mutations. Mice homozygously lacking dystroglycan show embryonic lethality, thought to arise from defects in extra-embryonic structures and their association with the extra-cellular matrix (Williamson).
The sarcoglycans are all N-glycosylated transmembrane proteins with a large extra-cellular domain containing a carboxyl-terminal cluster containing several cysteine residues, a single transmembrane region and a short intra-cellular domain. Both alpha/epsilon-sarcoglycan (SGCA/SGCE) and gamma/delta-sarcoglycan (SGCG/SGCD) are closely related proteins which are similar in size and sequence. gamma- and delta-sarcoglycan also show significant homology to beta- and zeta-sarcoglycan. SGCE-expression is both earlier and more widely than SGCA (Ettinger). The distribution of expression of SGCA and SGCE is also markedly different, with SGCA largely restricted to striated (cardiac and skeletal) muscle and very low levels in lung and none in brain (Ettinger). Althuogh alpha/epsilon-sarcoglycan and beta/gamma/delta-sarcoglycan have a related structure, their sequences differs greatly.
The sarcoglycans form a complex of glycoproteins, integrated in the membrane, which is fixed to the dystrophin axis by a lateral association with the dystroglycan complex. The sarcolgycan constituents of the DGC differ between different tissues. The smooth muscle DGC contains dystrophin, alpha- and beta-dystroglycan, beta-, delta- and epsilon-sarcoglycan and sarcospan. Furthermore, in smooth muscle, the molecular weight of delta-sarcoglycan appears to be slightly lower (Straub). Shi et al. (ASHG2002 abstract 940, Am.J.Hum.Genet. 71: S333) reported that the complex is generated by assembly of the sarcoglycans in a specific order. Assembly is initiated by SGCB to which SGCD binds. Next, SGCG binds to the complex at SGCD and finally SGCA attaches to SGCG.
Since alpha- and gamma-sarcoglycan are not expressed in smooth muscle cells and epsilon-sarcoglycan is not an integral component of the skeletal muscle DGC, the association between the sarcoglycan-complex and alhpa-dystroglycan is either mediated by beta- and delta-sarcoglycan or by sarcospan (Straub). Similarly, since homozygous beta-sarcoglycan missense mutations result in the total absence of alpha-, beta- and gamma-sarcoglycan from the skeletal muscle membrane, it has also been suggested that beta-sarcoglycan plays a critical role in the assembly and/or maintenance of the sarcoglycan complex (B—nnemann).
| Protein | striated muscle (cardiac, skeletal) |
smooth muscle | other tissues |
|---|---|---|---|
| SGCA | expressed | not expressed | not expressed |
| SGCB | selectively expressed | expressed (occurs post-nataly) |
detectable |
| SGCG | expressed | not expressed | not expressed |
| SGCD | selectively expressed | expressed | detectable |
| SGCE | detectable (high in embryo) |
expressed | widely expressed (early embryonic) |
| SGCZ |
Legend:
Sarcoglycan expression in adult tissues (data mainly from Straub).
Sgca is not detectable in smooth muscle but shows increasing expression in
striated muscle. Sgce expression is high throughout muscle development but
declines in striated muscle.
The syntrophins and dystrobrevin form a peripheral cytoplasmic 'subcomplex' thought to serve as a scaffold for signalling proteins. .he syntrophins (a, b1, b2) and a-dystrobrevin are excellent candidates as primary genetic causes for human muscle disease. They are associated with dystrophin at the sarcolemma, and the syntrophins and dystrobrevin are secondarily abnormal in Duchenne muscular dystrophy [30,32]. Knockout mouse models for both a-syntrophin, and a-dystrobrevin show abnormalities which will potentially correlate with a human muscle disease phenotype [42,43]. However, to date, no primary abnormality of these proteins has been described in human skeletal muscle disease. The aims of our study are to characterise the immunohistochemical expression of syntrophins and dystrobrevin in normal human muscle, and in patients in whom the underlying genetic cause of their muscle disease remains unknown.
The syntrophins are a heterogeneous multigene family of modular, 59 kDa cytoplasmic, phosphorylated, adaptor proteins sharing a common domain structure. Syntrophins comprise five different proteins; one alpha (alpha-1, acidic), two beta, (beta-1 and beta-2, basic) and two gamma (gamma-1 and gamma-2). Ahn et al. have reported the existence of a syntrophin-like protein (SNTL - EST25263), which turned out to be the human homologue of mouse syntrophin-beta2. alpha-Syntrophin is the major isoform of adult mouse skeletal muscle. In mouse, alpha and beta1-syntrophin are localised in the sarcolemma and the neuromuscular junction (NMJ). gamma2-Syntrophin is reportedly found at the sarcolemma, but seems not localised to the NMJ [13], while beta2-syntrophin is largely confined to the NMJ, with occasional presence at the sarcolemma.
The syntrophins (alpha, beta1 and beta2) bind directly to members of the dystrophin protein family: dystrophin, utrophin and a-dystrobrevin. The syntrophin complex binds to the second half of the carboxyl-terminal domain of dystrophin (the S-domain). alpha-syntrophin binds to the 3'-half of exon 73 and the 5'-half of exon 74 (i.e. amino acids 3444-3494). beta1-syntrophin and/or A0 bind to the 3'-half of exon 74 and the 5'-half of exon 75. The beta1-syntrophin binding site is restricted to exon 74. Syntrophin also associates with neuronal nitric oxide synthase (nNOS).
Based on their domain structure and association with neuronal nitric oxide synthase (nNOS, Brenman et al. [1996]), aquaporin-4 (Adams et al., 2001), ion channels (Gee et al., 1998), and kinases (Hogan et al., 2001) syntrophins are thought to function as modular adaptors that recruit signalling proteins to the membrane via association with the DGC.
The dystrobrevins alpha (DTNA) and beta (DTNB) are members of the dystrophin protein family encoded by two different genes. The mammalian dystrobrevin genes encode several protein isoforms that are expressed in different tissues, including brain and muscle. Blake et al. designated the isoform expressed in muscle as alpha-dystrobrevin (DTNA) and used the designation beta-dystrobrevin (DTNB) for the dystrophin-related protein abundantly expressed in brain and other tissues but not in muscle.
Blake found that the composition of the dystrophin-associated protein complex in the brain differs from that in muscle. Because beta-dystrobrevin and dystrophin are expressed in similar populations of neurons in the hippocampus and cortex, it is possible that beta-dystrobrevin interacts directly with dystrophin. If this is the case, then beta-dystrobrevin levels may be reduced in DMD patients similar to the reduction in sarcolemmal staining seen with other components of the DGC in dystrophic muscle. The findings may be relevant to the cognitive dysfunction affecting DMD/BMD patients.
Sarcospan (SSPN, Crosbie), originally described as 25DAP / A5 (Ervasti, Yoshida), was the last protein of the DGC that could be characterized at the molecular level. Cloning of the gene was hampered mainly by the difficulty to raise antibodies against the purified protein (rabbit protein injected in sheep and goat). Crosbie et al. purified 25DAP / A5 from rabbit skeletal muscle, micro-sequenced two Lys-C digestion products and detected a database homology with one peptide of the human and mouse Kirsten ras-associated gene (KRAG). The sarcospan gene maps to chromosome 12p11.2, overlapping the locus for CFEOM (congenital fibrosis of the extraocluar muscle) locus, mapped at 12p11.2-q12. Sarcospan encodes a major 6.5 kb transcript present exclusively in skeletal and cardiac muscle, and a 4.5 kb transcript in many other tissues. The primary sarcospan amino acid sequence predicts four transmembrane spanning domains and consensus phosphorylation sites for cAMP-dependent protein kinase, protein kinase C and casein kinase II. Overall sarcospan topology is similar to that of the tetraspan proteins.
Caveolin is a small molecular weight protein shown to be associated with the caveolar plasma membranes. Caveolin-3, or M-caveolin, was identified as a muscle-specific form of the caveolin family (Way, Tang). In skeletal muscle, caveolin-3 is localized to the sarcolemma, coinciding with dystrophin. Some caveolin-3 co-purifies with the dystrophin-associated glycoprotein complex and can be immunoprecipitated with dystrophin antibodies (Song, Ervasti, McNally), which suggests the existence of a discrete protein complex containing both proteins. However, Crosbie showed that CAV3 is not an integral member of the DGC, since its association is not as strong as that of other members. The number and size of caveolae are abnormal in DMD-patients. Caveolin-3 directly interacts with neuronal nitric oxide synthase (nNOS) and binding results in the loss of NOS activity (Venema...).
Mutations in the proteins of the sarcoglycan complex appear to be a significant cause of recessive autosomally inherited muscular dystrophy. The phenotypes range from closely resembling Duchenne (DLMD = Duchenne-like muscular dystrophy / SCARMD = severe childhood autosomal recessive muscular dystrophy) to the late-onset forms of Limb-Girdle muscular dystrophy (LGMD) Together, these diseases are indicated as the "sarcoglycanopathies". The differences in the expression of the sarcolgycans, especially those between SGCA/SGCC versus SGCB/SGCD (where smooth muscle seems unaffected), have been suggested to cause clinical differences. Straub et al. suggest that, based on the differences in sarcoglycan expression, analysis of skin biopsies might be helpful in the diagnosis of LGMD.
Mutations in caveolin-3 have been found in patients with an autosomal dominant form of Limb-Girdle muscular dystrophy (type 1C).
A tabular summary of all the proteins mentioned and the muscular dystrophies involved, with links to OMIM, GenBank, MEDLINE, etc. can be found at the "Muscular Dystrophies" page.
| Top of page | the DGC in
Invertebrates |
| Sarcoglycan homepage | Dystroglycan
homepage | Muscular dystrophies |
| LMDp homepage | Remarks /
information | Copyrightˋ, liability
|
{kind=link}
{kind=link}