(last modified September, 2008)
The DMD (dystrophin) sequence variants present in the DMD sequence variation database and the DMD deletion and duplication database are a collection of all variants we have found in literature and sequence databases, extended with those submitted directly to the database (i.e. not yet published). The DMD sequence variation database contains all variations that have been characterized at the nucleotide level, i.e. including deletions, duplications and translocations of which the breakpoints have been sequenced. The DMD deletion and duplication database contains all deletion and duplication mutations that have not been characterised at the nucleotide level; these changes were analysed e.g. using multiplex-PCR, Southern blotting, MAPH or MLPA.
Selected lists of the variations are available for the categories;
When you notice that we have missed variations or when you detect mistakes, tell us immediately. We would like to have the databases as complete, error-free and up-to-date as possible. When you have detected variations, please let us know and submit them electronically. Please note that we collect ALL changes, including those of uncertain pathogenicity and those identified before in other patients; the database should also contain information regarding the frequency in which mutations are found. When you have large list in electronic format, e.g. text-editor or spreadsheet, we would be happy tot assist you with uploading.
An important, often neglected type of variants is those of which it is not yet clear whether they are disease-causing or not (unclassified variant). The problem in such cases is that the sequence change detected nearly always segregates with the disease allele. Although analysis of up to 100 or more normal chromosomes may not reveal the same variation, this does not necessarily mean that the change is disease-causing; it might as well represent a rare neutral variant (polymorphism). Because of the uncertainty, the unclassified nature of the change, it is mostly not reported to the database and it is forgotten. However, unclassified variants are most important and should be reported immediately. One of the most powerful aspects of an internet-based database as a central repository is that the identification of these unclassified variants can be spread quickly and world-wide. Everybody who finds the same change has then the possibility to help sorting out the issue "pathogenic or not ?". Identified in a healthy control points to a non-deleterious change, identification in a patient supports a disease-causing nature. YOU are requested to contact us, and the reporting researcher, immediately when you find a similar sequence variant. Thus, do not forget to report ALL variants encountered in healthy controls as well as all changes of which you are not yet certain. The longer the list of variants identified, the more informative it will be.
Sequence changes in the DMD gene can also be found in some other databases;
To assist in the discrimination between disease-causing and not disease-causing amino acid substitutions we are preparing protein-derived sequence alignments. These alignements include dystrophins from other species, dystrophin-related proteins (e.g. utrophin, DRP2, dystrobrevin, etc.), alignements of the individual repeats from the central rod domain and alignments with proteins which show homology with specific dystrophin domains (e.g. actin, spectrin, etc.).
The DMD mutation data collected thus far show clear differences regarding their frequency, distribution and parental origin of the different types of mutations, i.e. deletion, duplication, point mutation.
Overview of the frequencies of specific types of mutations found in DMD-patients as reported by several studies. Type of change: the type of change identified, split to the three respective basic levels, DNA, RNA and protein. Studies reporting the data: Tuffery-Giraud et al. (2004) using PTT, Hofstra et al. (2004) using DGGE, Mendell et al. (2001) / Buzin (2005) using SSCA (DOVAM) and Flanigan et al. (2003) using direct sequencing (SCAIP).
| Type of change | Tuffery-Giraud
(2004) and pers.comm. (PTT) |
Hofstra
(2004) (DGGE) |
Buzin
(2005) (SSCA) |
Flanigan
(2003) (sequencing) |
Hamed
et al. 2006 (cDNA sequencing) |
|---|---|---|---|---|---|
| TOTAL reported | 89 | ? | ? | ? | 15 |
| no change identified | 3* | ? | ? | ? | 5 |
| on DNA - level | 86 | 35 | 127 | 17 | 10 |
| substitution | 45 (52%) | 22 (63%) | 56 (44%) | 14 | 5 |
| deletion (small) | 18 (21%) | 8 (23%) | 28 (22%) | 2 | 3 |
| deletion exon(s) | 8 (9%) *1 not detected by PTT |
- | 10 (8%) | - | - |
| duplication (small) | 1 (1%) | 4 (11%) | 9 (7%) | - | - |
| duplication exon(s) | 12 (14%) *6 not detected by PTT |
- | 18 (14%) | - | 3 |
| insertion (small) | 1 (1%) | - | 1 (1%) | 1 | - |
| deletion/insertion (indel) | 1 (1%) | 1 (3%) | 5 (4%) | - | - |
| complex / other | - | - | - | ||
| on RNA - level | |||||
| uncertain | - | - | 1 (1%) | - | - |
| affecting splice site | 16 (19%) | 8 (23%) | 8 (6%) | 2 | - |
not affecting splice site |
70 (81%) | 27 (77%) | 118 (93%) | 15 | 10 |
| on protein - level | |||||
| uncertain | - | - | 1 (1%) | - | - |
| missense | - | - | - | 2 | - |
| no start (change ATG) | - | - | - | - | - |
| nonsense | 31 (36%) | 14 (40%) | 48 (36%) | 10 | 5 |
| no stop | - | - | - | - | - |
| frame shift | 52 (60%) incl. 5 (multi-)exon del, 12 (multi-)exon dup |
21 (60%) | 75 (59%) | 5 | 2 |
| deletion (no frame shift) | 3 (3%) | - | 1 (1%) | - | - |
| duplication (no frame shift) | - | - | 2 (2%) | - | 3 |
| insertion (no frame shift) | - | - | - | - | - |
*updated after personal contact with the author.
......
Duchenne muscular dystrophy (DMD) is a recessive X linked disorder with an incidence of 1 in 3,500 live born males. The incidence of BMD is much lower, about 1 in 18,500. In 1/3 of the cases the disease is the result of a de novo mutation in the DMD gene, i.e. the mutation is detected in the patient's DNA (isolated from blood) and not in the patient's mother's DNA (isolated from blood). In 1987 Bakker et al. published two DMD families with an apparent germline mosaicism, i.e. a somatic non-carrier mother transmits a de novo mutation more then once (consequently several of her germ cells carry the mutation). Based on empirical data the DMD/BMD recurrence risk for mothers of a patient carrying a de novo mutation was estimated to lie between 7 and 10 % (Bakker 1989; van Essen 1992) when the risk haplotype is unknown.
Familial and sporadic cases and recurrence risk
Several studies have focused on the parental origin of the different types of sequence variants found in the DMD gene, i.e. deletion, duplication, point mutation. Passos-Bueno et al. (1992) based their study on the observation that among proven germ line mosaic cases a higher frequency of proximal deletions were found compared to that expected; observed 78% (18/23), expected ~25% (Table I).
| Mosaic cases | Observed | Expected |
|---|---|---|
| proximal deletion | 18 | 1 |
| distal deletion | 5 | 3 |
| Total | 23 | 4 |
Table I - location of deletions in the DMD gene reported for 23 germ line and somatic mosaic cases (from Passos-Bueno et al., 1992). In addition, one proximal duplication was reported.
The frequency of germ line mosaicism has a direct effect on "de novo" mutations being multiply transmitted, increasing their chance of becoming "familial". These findings predict a difference in the proportion of proximal versus distal deletions between familial and isolated cases. When the distribution of deletions was compared between isolated (proxomal:distal = 1:3) and familial cases (1:1, Table II), the predicted difference was indeed found.
| Deletions | Brazil | Nederland | Together | Total | |||
|---|---|---|---|---|---|---|---|
| location | proximal | distal | proximal | distal | proximal | distal | |
| familial cases | 10 | 8 | 16 | 21 | 26 (47%) |
29 (53%) |
55 (24%) |
| isolated cases | 28 | 63 | 22 | 66 | 50 (28%) |
129 (72%) |
179 (76%) |
| Total | 38 | 71 | 38 | 87 | 76 (32%) |
158 (68%) |
234 |
Table II - analysis of 473 unrelated DMD patients from two centers, 254 from Sao Paulo (Brazil) and 219 from Leiden (Nederland). One-generation families were excluded from the study since they could harbor hidden familial cases as well as germ line mosaic cases and de novo mutations in the mother's parents.
Bakker (1989), Passos-Beuno (1990) and van Essen (1992) determined that the recurrence risk in DMD cases with a proven de novo mutation is 7-9% (14-18% when the risk haplotype is known). In 45 of the Leiden families containing a proven de novo mutation (20 proximal and 25 distal) 7 cases of mosaicism were identified (6 proximal, 1 distal; Passos-Bueno (1992) In the 45 families the at-risk haplotype was transmitted 44 times, 7 times resulting in affected and 37 times in normal subjects. Proximal mutations occured 6 of 22 times (30%) and distal mutations 1 of 22 times (4%). These observations have important consequences for these recurrence-risk estimates. Based on the location of the deletion and the at-risk haplotype known, Passos-Bueno (1992) calculated an increased recurrence risk for de novo proximal deletions (~30%) compared to de novo distal deletions (~4%).
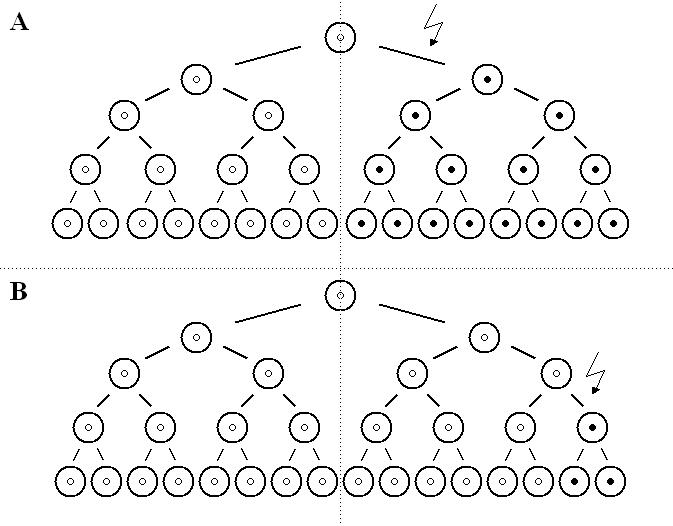
The data indicate that the origin of deletions in the proximal and distal hot spots differ (Fig.1).
Figure 1
A. proximal de novo deletions in the DMD gene occur early in embryonic development,
more cells are affected, the risk of passing the mutation to the next generation
(recurrence risk) increases and familial cases result. B. distal de novo deletions
occur later (mitotically), fewer cells are affected, the recurrence risk decreases and
generally only isolated cases result.
When proximal deletions occur very early in embryogenesis they are probably not restricted to the germ line. If so, low levels of somatic mosaicism might be detectable in mothers of de novo patients and this level might be predictive for the recurrence risk (Passos-Bueno 1992).
Deletions, duplications and point mutations
Analysing 280 DMD families, Grimm et al (1994) showed evidence that point mutations generally have a grandpaternal origin, presumably arising from errors during spermatogenesis (in 24 of 37 sporadic cases the presumed point mutation was on the grandpaternal haplotype). In contrast, deletions in general had a grandmaternal origin (26/42 cases).
At the WMS2005 meeting in Iguassu (Brazil) Helderman et al. (Leiden, Nederland) reported to have diagnostically tested about 1,500 DMD/BMD patients/families and identified the pathogenic mutation in most of them. To determine the recurrence risk because of germline mosaicism 318 families could be used; the risk haplotype without mutation was transmitted 192,5 times and with mutation for a second time in 19 cases. The recurrence risk when transmitting the risk haplotype was thus calculated to be 9% (19/[192,5 +19] with 5,1-12,8% 95% confidence interval). In 301 families the origin of the de novo mutation could be determined (Table III).
| Mutation type | Maternal | Grandmaternal | Grandpaternal | Total |
|---|---|---|---|---|
| deletion | 183 | 37 | 15 | 235 |
| duplication | 25 | 3 | 5 | 33 |
| point mutation | 23 | 3 | 6 | 32 |
| unknown | 1 | 0 | 0 | 1 |
Total |
232 | 43 | 26 | 301 |
Table III - origin of the de novo mutation in 301 families as determined by Helderman et al. (Leiden, Nederland).
Overall no difference in sex ratio could be determined for the origin of de novo mutations. The data do show that duplications and point mutations more frequently have a paternal origin. Helderman also report that duplications overall have a more proximal location, quite different from the bi-modal distribution of deletions.
| Top of page | LMDp home page
|
| Diseases on these pages | Gene / disease
table | Muscular Dystrophy databases |
| Remarks / information | Copyright©,
liability |